Extracting Embeddings with scConcept#
This tutorial demonstrates how to extract embeddings from single-cell RNA-seq data using scConcept.
import os
from pathlib import Path
import scanpy as sc
from concept import scConcept
The directory where the pre-trained model will be downloaded:
cache_dir = Path("./cache/")
os.makedirs(cache_dir, exist_ok=True)
Download a sample dataset:
filename = cache_dir / "multiome_gex_processed_training.h5ad"
url = "https://openproblems-bio.s3.amazonaws.com/public/explore/multiome/multiome_gex_processed_training.h5ad"
if not os.path.exists(filename):
import urllib.request
print(f"Downloading {filename} ...")
urllib.request.urlretrieve(url, filename)
else:
print(f"{filename} already exists, skipping download.")
adata = sc.read(filename)
print(adata)
Downloading cache/multiome_gex_processed_training.h5ad ...
AnnData object with n_obs × n_vars = 42492 × 13431
obs: 'pct_counts_mt', 'n_counts', 'n_genes', 'size_factors', 'phase', 'cell_type', 'pseudotime_order_GEX', 'batch', 'pseudotime_order_ATAC', 'is_train'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dataset_id', 'organism'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
Load a pre-trained scConcept model:
concept = scConcept(cache_dir=cache_dir)
concept.load_config_and_model(model_name='corpus40M-model30M')
Extract embeddings:
Indicate the column name of the gene ids in the adata.var of the format: ENSGXXXXXXXXX
result = concept.extract_embeddings(
adata=adata,
batch_size=64, # Adjust batch size based on your GPU memory
gene_id_column="gene_ids",
)
adata.obsm['X_scConcept'] = result['cls_cell_emb']
print(f"CLS embeddings: {adata.obsm['X_scConcept'].shape}")
CLS embeddings: (42492, 512)
Compute UMAP on the embeddings:
sc.pp.neighbors(adata, use_rep='X_scConcept')
sc.tl.umap(adata)
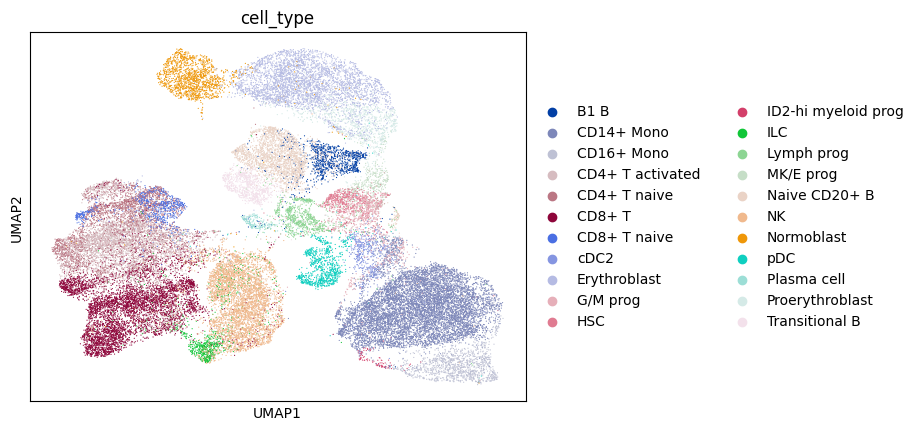
sc.pl.umap(adata, color='cell_type')