Estimating Mutual Information with scConcept#
This tutorial demonstrates how to estimate mutual information between a gene panel and the full transcriptome using scConcept.
import os
from pathlib import Path
import numpy as np
import scanpy as sc
from concept import scConcept
The directory where the pre-trained model and example dataset will be stored:
cache_dir = Path("./cache/")
os.makedirs(cache_dir, exist_ok=True)
Download the same sample dataset used in the embedding extraction tutorial:
filename = cache_dir / "multiome_gex_processed_training.h5ad"
url = "https://openproblems-bio.s3.amazonaws.com/public/explore/multiome/multiome_gex_processed_training.h5ad"
if not os.path.exists(filename):
import urllib.request
print(f"Downloading {filename} ...")
urllib.request.urlretrieve(url, filename)
else:
print(f"{filename} already exists, skipping download.")
adata = sc.read(filename)
print(adata)
cache/multiome_gex_processed_training.h5ad already exists, skipping download.
AnnData object with n_obs × n_vars = 42492 × 13431
obs: 'pct_counts_mt', 'n_counts', 'n_genes', 'size_factors', 'phase', 'cell_type', 'pseudotime_order_GEX', 'batch', 'pseudotime_order_ATAC', 'is_train'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dataset_id', 'organism'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
Load a pre-trained scConcept model:
concept = scConcept(cache_dir=cache_dir)
concept.load_config_and_model(model_name="corpus40M-model30M")
Subsample cells for a faster example, then define a panel. Here we use the first 200 genes from adata.var['gene_ids'] only as a simple example. In practice, replace panel_gene_ids with the gene IDs from your panel.
estimate_size = min(2048, adata.n_obs)
rng = np.random.default_rng(seed=42)
random_indices = rng.choice(adata.n_obs, estimate_size, replace=False)
adata_subset = adata[random_indices].copy()
panel_gene_ids = adata_subset.var["gene_ids"].astype(str).to_numpy()[:200]
panel_mask = adata_subset.var["gene_ids"].astype(str).isin(panel_gene_ids).to_numpy()
adata_panel = adata_subset[:, panel_mask].copy()
print(f"Cells used: {adata_subset.n_obs}")
print(f"Full transcriptome genes: {adata_subset.n_vars}")
print(f"Panel genes used: {adata_panel.n_vars}")
Cells used: 2048
Full transcriptome genes: 13431
Panel genes used: 200
Extract embeddings for the full transcriptome and for the panel, then estimate mutual information between them:
embedding_kwargs = {
"batch_size": 64,
"gene_id_column": "gene_ids",
"return_type": "torch",
}
cell_embs_full = concept.extract_embeddings(adata_subset, **embedding_kwargs)["cls_cell_emb"]
cell_embs_panel = concept.extract_embeddings(adata_panel, **embedding_kwargs)["cls_cell_emb"]
mutual_info = concept.estimate_mutual_information(cell_embs_full, cell_embs_panel)
print(f"Estimated mutual information: {mutual_info:.4f}")
Estimated mutual information: 0.7974
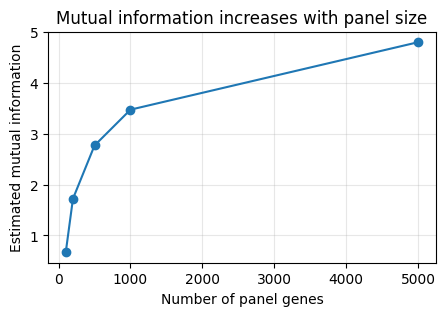
Larger Panels Increase Mutual Information#
This simple example uses nested panels of increasing size and compares each one to the same full-transcriptome embedding.
import matplotlib.pyplot as plt
mean_expression = np.asarray(adata_subset.X.mean(axis=0)).ravel()
gene_order = adata_subset.var.iloc[np.argsort(mean_expression)[::-1]]["gene_ids"].astype(str).to_numpy()
panel_sizes = [100, 200, 500, 1000, 5000]
panel_sizes = [size for size in panel_sizes if size <= len(gene_order)]
mutual_infos = []
for panel_size in panel_sizes:
panel_gene_ids = gene_order[:panel_size]
panel_mask = adata_subset.var["gene_ids"].astype(str).isin(panel_gene_ids).to_numpy()
adata_panel = adata_subset[:, panel_mask].copy()
cell_embs_panel = concept.extract_embeddings(adata_panel, **embedding_kwargs)["cls_cell_emb"]
mutual_info = concept.estimate_mutual_information(cell_embs_full, cell_embs_panel)
mutual_infos.append(mutual_info)
plt.figure(figsize=(5, 3))
plt.plot(panel_sizes, mutual_infos, marker="o")
plt.xlabel("Number of panel genes")
plt.ylabel("Estimated mutual information")
plt.title("Mutual information increases with panel size")
plt.grid(True, alpha=0.3)
plt.show()